
Lab Journal Club
Fostering deep scholarly insight by critically reviewing research papers and unearthing innovative ideas!
The Yoo Lab journal club meets on Fridays at 10:30 AM, usually in the MPR meeting room.
Every two weeks, each lab member presents an article they have recently read. We introduce the research group behind the paper and discuss its key questions, hypotheses, and findings in (sometimes painstaking) detail. To wrap up, each member briefly shares their own take on the paper they prepared.

[2026.06.26] Exercise alleviates cognitive dysfunction in Alzheimer's disease mice via skeletal muscle-derived extracellular vesicles that enhance plaque clearance by microglia. Lin et al., Nat Aging. 2026
Exercise confers cognitive benefits in Alzheimer's disease (AD), yet the underlying mechanisms remain incompletely understood. Skeletal muscle functions as an endocrine organ that secretes myokines which affect the homeostasis of extra-muscular organs, including the brain. Here we found that swimming exercise promotes secretion of skeletal muscle-derived extracellular vesicles (SKM-EVs), which are subsequently taken up via pinocytosis by microglia. Gain-of-function and loss-of-function experiments showed that exercise-induced SKM-EVs induce polarization of disease-associated microglia and enhance the clearance of amyloid-beta plaques. Furthermore, miR-378a-3p was identified as a key microRNA cargo in SKM-EVs, regulating lipid metabolism in disease-associated microglia by targeting p110α. Importantly, administration of extracellular vesicles derived from miR-378a-overexpressing myotubes alleviated cognitive impairment in AD mice. Together, our findings demonstrate that exercise-induced SKM-EVs could serve as a myokine, mediating communication from skeletal muscle to the brain, providing a potential exercise-mimicking therapeutic strategy for AD.
Research group: Baosheng Guo [Link] / Pubmed: [Link] / Speaker: Alex
Rating: ★★★
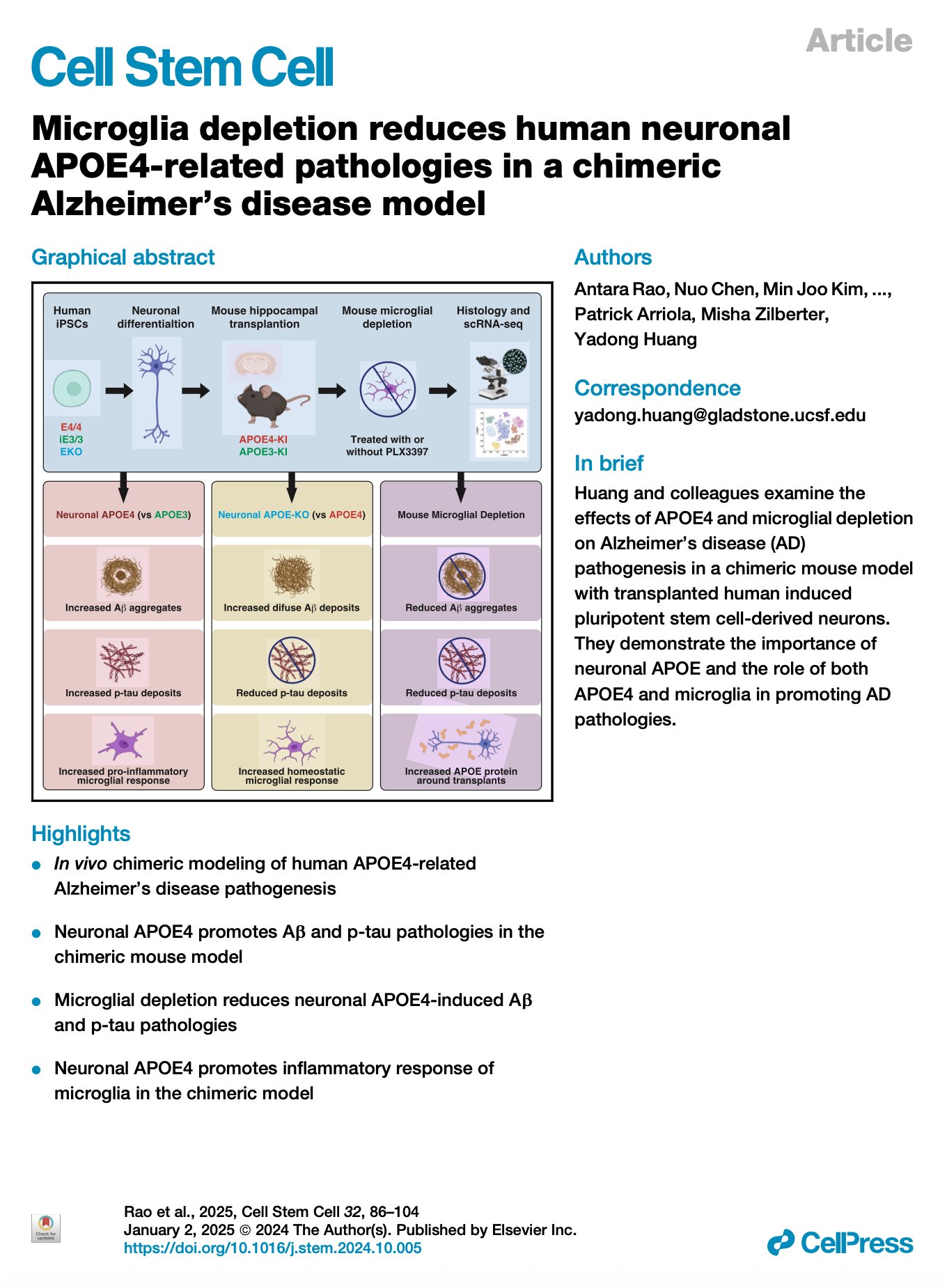
[2026.06.12] Microglia depletion reduces human neuronal APOE4-related pathologies in a chimeric Alzheimer's disease model. Rao et al., Cell Stem Cell. 2025
Despite strong evidence supporting the important roles of both apolipoprotein E4 (APOE4) and microglia in Alzheimer's disease (AD) pathogenesis, the effects of microglia on neuronal APOE4-related AD pathogenesis remain elusive. To examine such effects, we utilized microglial depletion in a chimeric model with induced pluripotent stem cell (iPSC)-derived human neurons in mouse hippocampus. Specifically, we transplanted homozygous APOE4, isogenic APOE3, and APOE-knockout (APOE-KO) iPSC-derived human neurons into the hippocampus of human APOE3 or APOE4 knockin mice and then depleted microglia in half of the chimeric mice. We found that both neuronal APOE and microglial presence were important for the formation of Aβ and tau pathologies in an APOE isoform-dependent manner (APOE4 > APOE3). Single-cell RNA sequencing analysis identified two pro-inflammatory microglial subtypes with elevated MHC-II gene expression enriched in chimeric mice with human APOE4 neuron transplants. These findings highlight the concerted roles of neuronal APOE, especially APOE4, and microglia in AD pathogenesis.
Research group: Yadong Huang [Link] / Pubmed: [Link] / Speaker: 김소정
Rating: ★☆☆

[2026.05.22] Microglia-mediated protection against Alzheimer's disease pathology and detrimental effects in white matter revealed by Ptpn6 deletion. Etxeberria et al., Neuron. 2026
Genetic variants affecting microglia can cause early-onset neurodegeneration or elevate Alzheimer's disease risk. To nominate regulators of relevant signaling pathways, we developed a genome-wide CRISPR screen in primary macrophages focused on survival. We identified Ptpn6, which encodes the inhibitory phosphatase SHP-1, as a crucial regulator for macrophage survival under reduced CSF1R signaling conditions in vitro. Deletion of Ptpn6 from adult microglia in vivo enhanced survival and decreased neuritic dystrophy around amyloid plaques in the TauPS2APP model of Alzheimer's disease. However, deletion also dysregulated homeostasis in normal white matter and exacerbated neurodegeneration in disease. Heterozygous deletion revealed a differential gene-dosage sensitivity for beneficial and detrimental effects, exhibiting reduced neuritic damage near plaques without white-matter harm. Single-cell RNA sequencing uncovered multiple disease-associated microglia (DAM)-like transcriptional states, with Lgals3+ microglia emerging alongside neurodegeneration after Ptpn6 deletion. In all, these findings reveal both the protective and latent degenerative potential of microglia held in check by Ptpn6.
Research group: Christopher J Bohlen Lab at Genetech / Pubmed: [Link] / Speaker: 김도현
Rating: ★★★
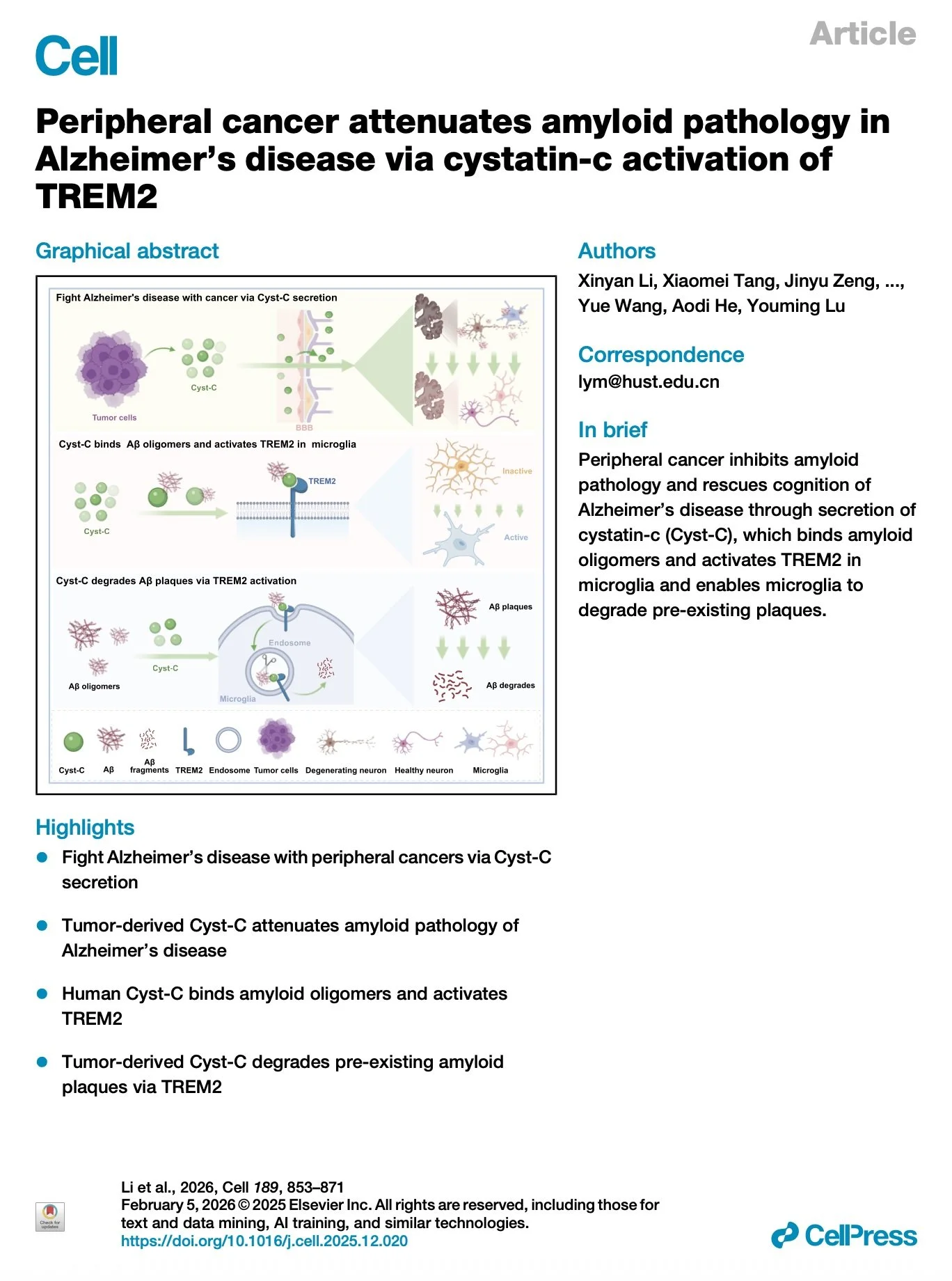
[2026.05.08] Peripheral cancer attenuates amyloid pathology in Alzheimer's disease via cystatin-c activation of TREM2. Li et al., Cell. 2026
Alzheimer's disease (AD) and cancer are among the most devastating diseases worldwide. Epidemiological data indicate that the incidence of AD significantly decreases in patients with a history of cancer. However, whether and how peripheral cancer may affect AD progression is yet to be studied. Here, we find that peripheral cancer inhibits amyloid pathology and rescues cognition via secretion of cystatin-c (Cyst-C), which binds amyloid oligomers and activates triggering receptor expressed on myeloid cells 2 (TREM2) in microglia, enabling microglia to degrade the pre-existing amyloid plaques in AD mice. These effects of Cyst-C are abolished by a cell-type-specific deletion (Cx3cr1TREM2-/-) or mutation of TREM2 (TREM2R47H) or Cyst-C (Cyst-CL68Q) in microglia. Together, these findings provide significant conceptual advances into cancer neuroscience and establish therapeutic avenues that are distinct from the present amyloid-lowering strategies, aiming at degrading the existing amyloid plaques for precision-targeted AD therapy.
Research group: Youming Lu Lab [Link] / Pubmed: [Link] / Speaker: 서정민
Rating: ★★★

[2026.04.03] Distinct origins and niches determine the cellular responsiveness of CNS macrophages after repopulation. Fliegauf et al., Nat Immunol. 2026
Nonparenchymal central nervous system (CNS)-associated macrophages (CAMs) mediate immune responses at brain boundaries. Perivascular and leptomeningeal CAMs are collectively termed subdural CAMs (sdCAMs). Both sdCAMs and juxtaneuronal microglia are derived from embryonic yolk sac precursors, long-living and maintain their populations through self-renewal. Following depletion, microglia autonomously repopulate from single surviving cells. In contrast, the course of sdCAM repopulation remains poorly understood. Here, by combining multilineage fate mapping, multiomic profiling and high-resolution imaging, we demonstrate divergent repopulation dynamics between sdCAMs and microglia. Unlike microglia, sdCAMs do not renew cell-autonomously, but become transiently accessible to CCR2+Ly6C+ monocyte engraftment after niche induction in an integrin-dependent manner. Moreover, replenished monocyte-derived sdCAMs remain transcriptomically, epigenetically and functionally distinct from their embryo-derived counterparts. Finally, we present a protocol enabling selective exchange of sdCAMs, modulating disease response without functionally affecting microglia. These new insights into CNS immune biology suggest new therapeutic avenues for neuroinflammatory and neurodegenerative diseases.
Research group: Marco Prinz Lab [Link] / Pubmed: [Link] / Speaker: 이진아
Rating: ★★★
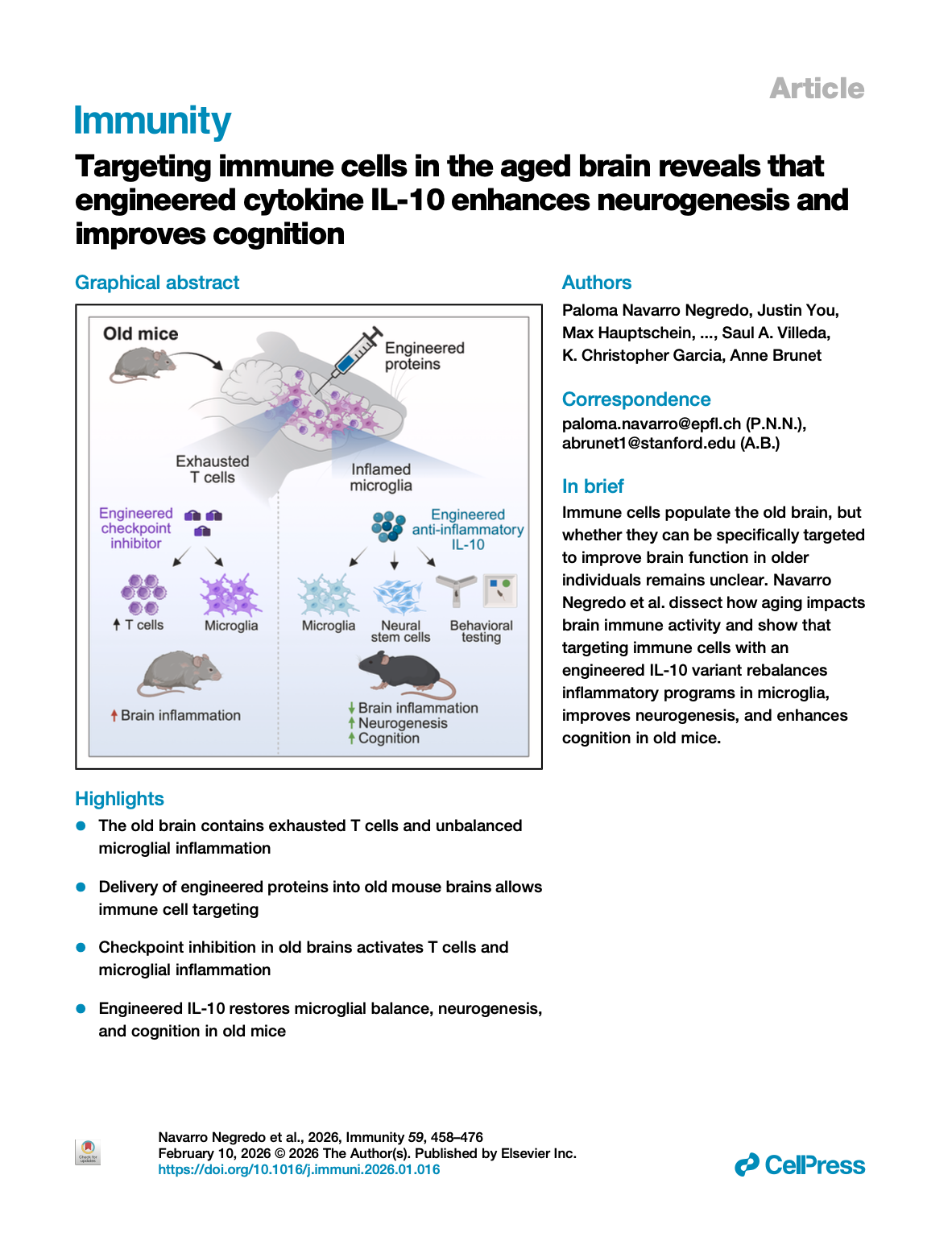
[2026.03.19] Targeting immune cells in the aged brain reveals that engineered cytokine IL-10 enhances neurogenesis and improves cognition. Negredo et al., Immunity. 2026
The immune system could play an important role in the age-related decline in brain function, yet specific immune-based strategies to enhance brain resilience in older individuals are lacking. Here, we combined engineered proteins and direct brain delivery to target immune cell populations within the old brain. We detected T cells with an exhaustion signature in the old brain and targeted them with a potent engineered checkpoint inhibitor (RIPR-PD1). This led to T cell expansion and strong pro-inflammatory responses in many brain cell types, notably microglia. To rescue age-related inflammatory imbalances in microglia, we used the anti-inflammatory cytokine interleukin (IL)-10. IL-10 boosted anti-inflammatory responses in old microglia, but it also triggered pro-inflammatory signaling. An engineered IL-10 variant that uncouples pro- and anti-inflammatory responses positively impacted the transcriptome of multiple cell types, enhanced neurogenesis, and improved cognition in aged mice. Our findings pave the way for immunotherapies for the aged brain.
Research group: Anne Brunet Lab [Link] / Pubmed: [Link] / Speaker: Alex
Rating: ★★★
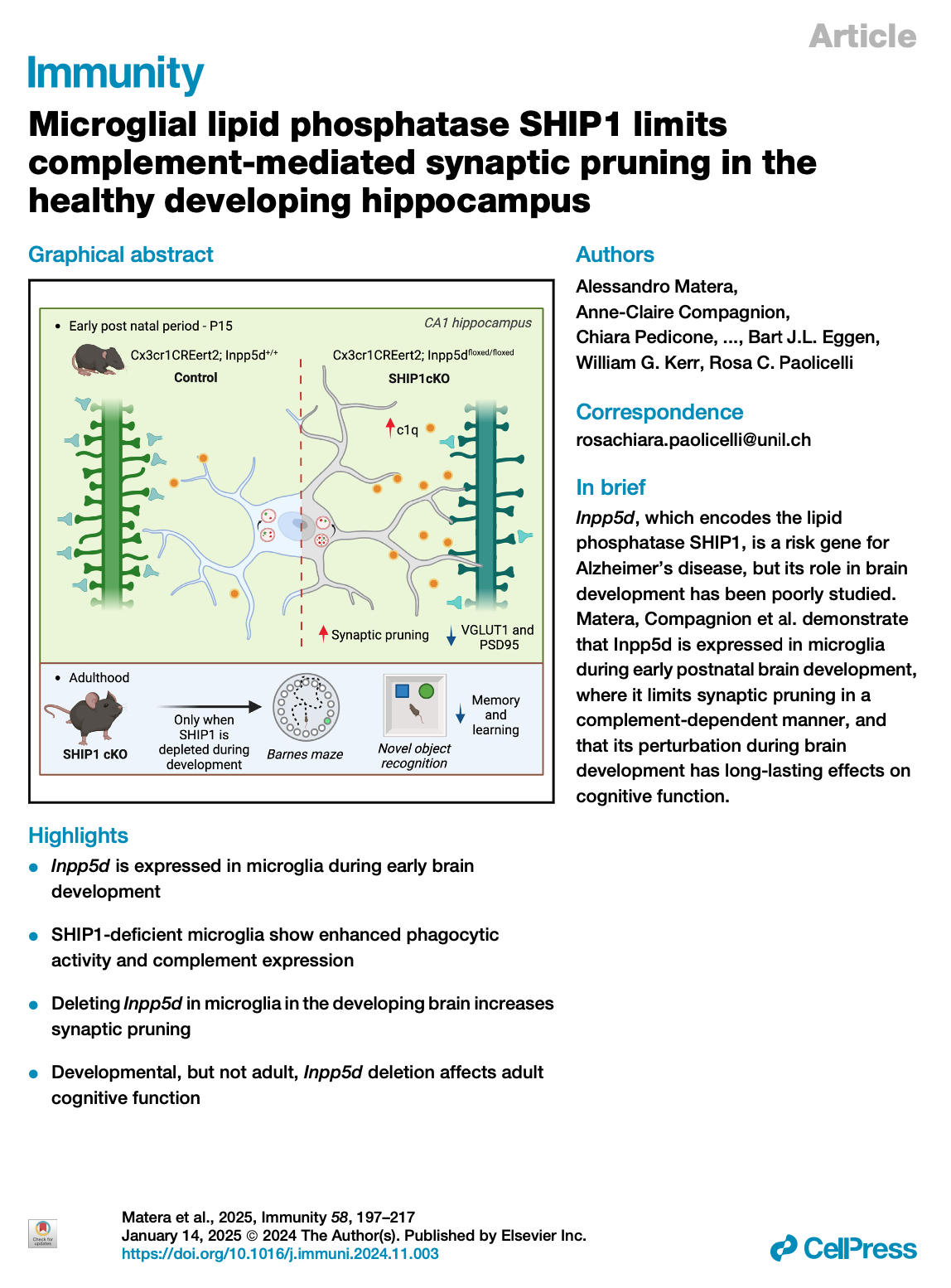
[2026.02.25] Microglial lipid phosphatase SHIP1 limits complement-mediated synaptic pruning in the healthy developing hippocampus. Albertini et al., Nat Neurosci. 2026
The gene inositol polyphosphate-5-phosphatase D (INPP5D), which encodes the lipid phosphatase SH2-containing inositol polyphosphate 5-phosphatase 1 (SHIP1), is associated with the risk of Alzheimer's disease (AD). How it influences microglial function and brain physiology is unclear. Here, we showed that SHIP1 was enriched in early stages of healthy brain development. By combining in vivo loss-of-function approaches and proteomics, we discovered that mice conditionally lacking microglial SHIP1 displayed increased complement and synapse loss in the early postnatal brain. SHIP1-deficient microglia showed altered transcriptional signatures and abnormal synaptic pruning that was dependent on the complement system. Mice exhibited cognitive defects in adulthood only when microglial SHIP1 was depleted early postnatally but not at later stages. Induced pluripotent stem cell (iPSC)-derived microglia lacking SHIP1 also showed increased engulfment of synaptic structures. These findings suggest that SHIP1 is essential for proper microglia-mediated synapse remodeling in the healthy developing brain. Disrupting this process has lasting behavioral effects and may be linked to vulnerability to neurodegeneration.
Research group: Rosa C Paolicelli Lab [Link] / Pubmed: [Link] / Speaker: 유용진
Rating: ★★☆

[2026.02.04] The Alzheimer's therapeutic Lecanemab attenuates Aβ pathology by inducing an amyloid-clearing program in microglia. Albertini et al., Nat Neurosci. 2026
Controversies over anti-amyloid immunotherapies underscore the need to elucidate their mechanisms of action. Here we demonstrate that Lecanemab, a leading anti-β-amyloid (Aβ) antibody, mediates amyloid clearance by activating microglial effector functions. Using a human microglia xenograft mouse model, we show that Lecanemab significantly reduces Aβ pathology and associated neuritic damage, while neither fragment crystallizable (Fc)-silenced Lecanemab nor microglia deficiency elicits this effect despite intact plaque binding. Single-cell RNA sequencing and spatial transcriptomic analyses reveal that Lecanemab induces a focused transcriptional program that enhances phagocytosis, lysosomal degradation, metabolic reprogramming, interferon γ genes and antigen presentation. Finally, we identify SPP1/osteopontin as a major factor induced by Lecanemab treatment and demonstrate its role in promoting Aβ clearance. These findings highlight that effective amyloid removal depends on the engagement of microglia through the Fc fragment, providing critical insights for optimizing anti-amyloid therapies in Alzheimer's disease.
Research group: Strooper Lab [Link] / Pubmed: [Link] / Speaker: 서정민
Rating: ★★☆

[2026.01.21] TGFβ signaling mediates microglial resilience to spatiotemporally restricted myelin degeneration. Zhu et al., Nat Neurosci. 2026
Microglia survey and regulate central nervous system myelination during embryonic development and adult homeostasis. However, whether microglia-myelin interactions are spatiotemporally regulated remains unexplored. Here, by examining spinal cord white matter tracts in mice, we determined that myelin degeneration was particularly prominent in the dorsal column (DC) during normal aging. This was accompanied by molecular and functional changes in DC microglia as well as an upregulation of transforming growth factor beta (TGF)β signaling. Disrupting TGFβ signaling in microglia led to unrestrained microglial responses and myelin loss in the DC, accompanied by neurological deficits exacerbated with aging. Single-nucleus RNA-sequencing analyses revealed the emergence of a TGFβ signaling-sensitive microglial subset and a disease-associated oligodendrocyte subset, both of which were spatially restricted to the DC. We further discovered that microglia rely on a TGFβ autocrine mechanism to prevent damage of myelin in the DC. These findings demonstrate that TGFβ signaling is crucial for maintaining microglial resilience to myelin degeneration in the DC during aging. This highlights a previously unresolved checkpoint mechanism of TGFβ signaling with regional specificity and spatially restricted microglia-oligodendrocyte interactions.
Research group: Lund Lab [Link] / Pubmed: [Link] / Speaker: 방지윤
Rating: ★★★

[2025.12.17] Microglia-neuron crosstalk through Hex-GM2-MGL2 maintains brain homeostasis. Frosch et al., Nature. 2025
As tissue-resident macrophages of the central nervous system parenchyma, microglia perform diverse essential functions during homeostasis and perturbations1. They primarily interact with neurons by means of synaptic engulfment and through the rapid elimination of apoptotic cells and non-functional synapses2. Here, by combining unbiased lipidomics and high-resolution spatial lipid imaging, deep single-cell transcriptome analysis and novel cell-type-specific mutants, we identified a previously unknown mode of microglial interaction with neurons. During homeostasis, microglia deliver the lysosomal enzyme β-hexosaminidase to neurons for the degradation of the ganglioside GM2 that is integral to maintaining cell membrane organization and function. Absence of Hexb, encoding the β subunit of β-hexosaminidase, in both mice and patients with neurodegenerative Sandhoff disease leads to a massive accumulation of GM2 derivatives in a characteristic spatiotemporal manner3. In mice, neuronal GM2 gangliosides subsequently engage the macrophage galactose-type lectin 2 receptor on microglia through N-acetylgalactosamine residues, leading to lethal neurodegeneration. Notably, replacement of microglia with peripherally derived microglia-like cells is able to break this degenerative cycle and fully restore central nervous system homeostasis. Our results reveal a mode of bidirectional microglia-neuron communication centred around GM2 ganglioside turnover, identify a microgliopathy and offer therapeutic avenues for these maladies.
Research group: Marco Prinz Lab [Link] / Pubmed: [Link] / Speaker: 이진아
Rating: ★★★
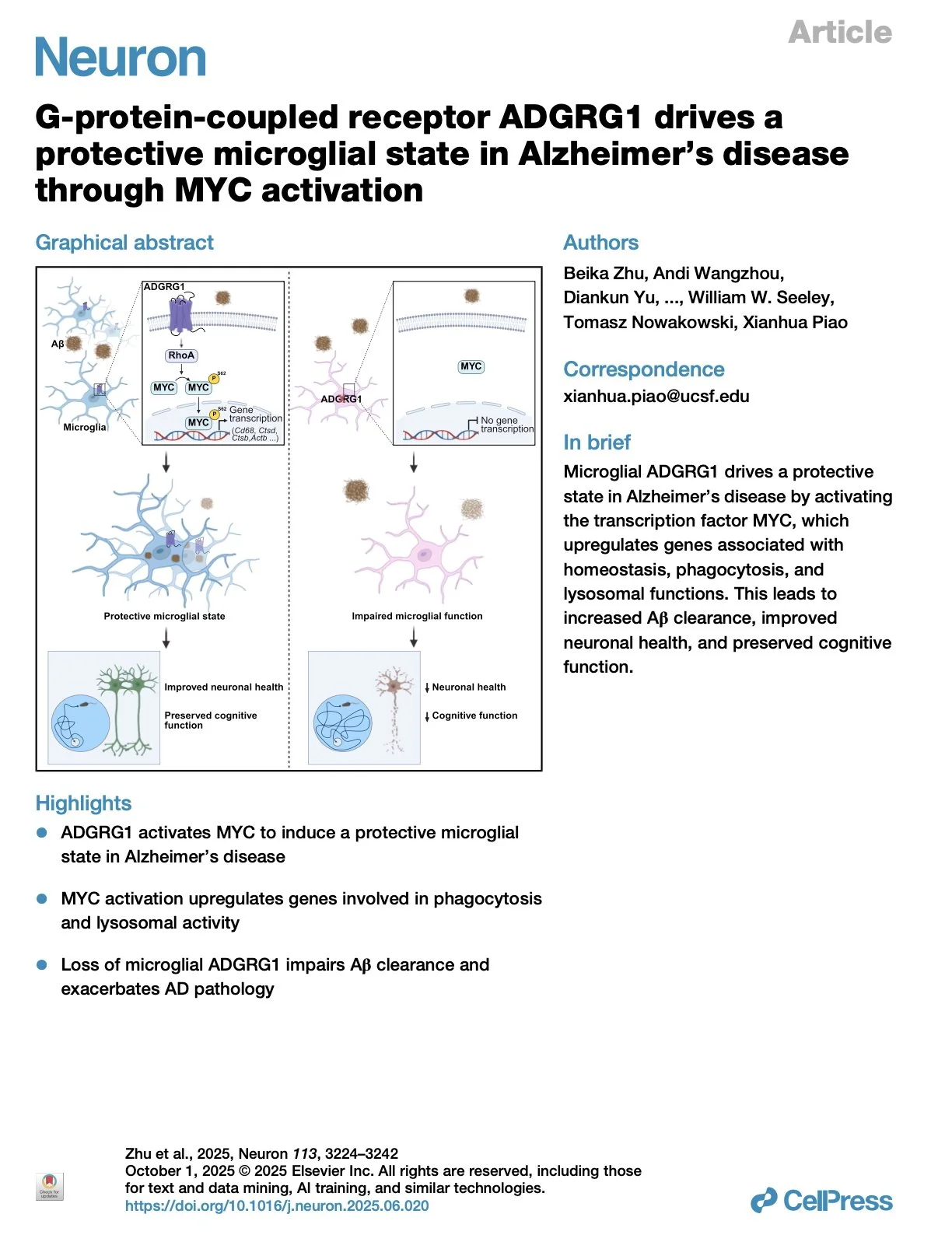
[2025.12.03] G-protein-coupled receptor ADGRG1 drives a protective microglial state in Alzheimer's disease through MYC activation. Zhu et al., Neuron. 2025
Germline genetic architecture of Alzheimer's disease (AD) indicates microglial mechanisms of disease susceptibility and outcomes. However, the mechanisms enabling protective microglial responses remain elusive. Here, we investigate the role of microglial ADGRG1, an adhesion G-protein-coupled receptor (aGPCR) specifically expressed in yolk-sac-derived microglia, in AD pathology using the 5xFAD mouse model. Transcriptomic analyses reveal that ADGRG1 activates the transcription factor MYC, leading to upregulation of genes involved in homeostasis, phagocytosis, and lysosomal functions, thereby promoting a protective microglial state. We demonstrate that deletion of Adgrg1 in microglia impairs MYC activation, resulting in increased amyloid-beta deposition, exacerbated neuronal loss, and cognitive deficits. Functional assays in mouse models and human embryonic stem cell-derived microglia confirm that ADGRG1 is required for Aβ phagocytosis. These findings uncover a GPCR-mediated pathway that drives a protective microglial state via MYC activation, suggesting potential therapeutic strategies to alleviate AD progression by enhancing microglial functional competence.
Research group: Xianhua Piao Lab [Link] / Pubmed: [Link] / Speaker: Alex
Rating: ★★☆ (Alex actually rated this paper 1.5 star.)

[2025.11.12] Transcriptional and epigenetic targets of MEF2C in human microglia contribute to cellular functions related to autism risk and age-related disease. Nguyen et al., Nat Immunol. 2025
MEF2C encodes a transcription factor that is critical in nervous system development. Here, to examine disease-associated functions of MEF2C in human microglia, we profiled microglia differentiated from isogenic MEF2C-haploinsufficient and MEF2C-knockout induced pluripotent stem cell lines. Complementary transcriptomic and functional analyses revealed that loss of MEF2C led to a hyperinflammatory phenotype with broad phagocytic impairment, lipid accumulation, lysosomal dysfunction and elevated basal inflammatory cytokine secretion. Genome-wide profiling of MEF2C-bound sites coupled with the active regulatory landscape enabled inference of its transcriptional functions and potential mechanisms for MEF2C-associated cellular functions. Transcriptomic and epigenetic approaches identified substantial overlap with idiopathic autism datasets, suggesting a broader role of human microglial MEF2C dysregulation in idiopathic autism. In a mouse xenotransplantation model, loss of MEF2C led to morphological, lysosomal and lipid abnormalities in human microglia in vivo. Together, these studies reveal mechanisms by which reduced microglial MEF2C could contribute to the development of neurological diseases.
Research group: Coufal Lab [Link] / Pubmed: [Link] / Speaker: 유용진
Rating: ★★☆

[2025.10.27] Young glial progenitor cells competitively replace aged and diseased human glia in the adult chimeric mouse brain. Vieira et al., Nat Biotechnol. 2024
Competition among adult brain cells has not been extensively researched. To investigate whether healthy glia can outcompete diseased human glia in the adult forebrain, we engrafted wild-type (WT) human glial progenitor cells (hGPCs) produced from human embryonic stem cells into the striata of adult mice that had been neonatally chimerized with mutant Huntingtin (mHTT)-expressing hGPCs. The WT hGPCs outcompeted and ultimately eliminated their human Huntington's disease (HD) counterparts, repopulating the host striata with healthy glia. Single-cell RNA sequencing revealed that WT hGPCs acquired a YAP1/MYC/E2F-defined dominant competitor phenotype upon interaction with the host HD glia. WT hGPCs also outcompeted older resident isogenic WT cells that had been transplanted neonatally, suggesting that competitive success depended primarily on the relative ages of competing populations, rather than on the presence of mHTT. These data indicate that aged and diseased human glia may be broadly replaced in adult brain by younger healthy hGPCs, suggesting a therapeutic strategy for the replacement of aged and diseased human glia.
Research group: Goldman Lab [Link] / Pubmed: [Link] / Speaker: 서정민
Rating: ★★☆

[2025.10.13] Iterative transcription factor screening enables rapid generation of microglia-like cells from human iPSC. Liu et al. Nat. Commun. 2025
Differentiation of induced pluripotent stem cells (iPSCs) into specialized cell types is essential for uncovering cell-type specific molecular mechanisms and interrogating cellular function. Transcription factor screens have enabled efficient production of a few cell types; however, engineering cell types that require complex transcription factor combinations remains challenging. Here, we report an iterative, high-throughput single-cell transcription factor screening method that enables the identification of transcription factor combinations for specialized cell differentiation, which we validated by differentiating human microglia-like cells. We found that the expression of six transcription factors, SPI1, CEBPA, FLI1, MEF2C, CEBPB, and IRF8, is sufficient to differentiate human iPSC into cells with transcriptional and functional similarity to primary human microglia within 4 days. Through this screening method, we also describe a novel computational method allowing the exploration of single-cell RNA sequencing data derived from transcription factor perturbation assays to construct causal gene regulatory networks for future cell fate engineering.
Research group: George M. Church Lab [Link] / Pubmed: [Link] / Speaker: 김민재

[2025.09.22] Identification of the velum interpositum as a meningeal-CNS route for myeloid cell trafficking into the brain. Hohsfield et al. Neuron. 2025
The borders of the central nervous system (CNS) host a repertoire of immune cells and mediate critical neuroimmune interactions, including the infiltration of peripheral myeloid cells into the CNS. Despite the fundamental role of leukocyte infiltration under physiological and pathological conditions, the neuroanatomical route of cell entry into the brain remains unclear. Here, we describe a specialized structure underneath the hippocampus, the velum interpositum (VI), that serves as a site for myeloid cell entry into the CNS. The VI functions as an extra-parenchymal leptomeningeal extension containing distinct myeloid cells subsets. Fate-mapping studies confirm meningeal and peripheral myeloid cell occupancy within the VI. Additionally, we highlight the distinct use of this route in the developing, irradiated, and demyelinating disease brain, indicating that myeloid cell trafficking through the VI could have important clinical implications for neurological disease.
Research group: Kim Green Lab [Link] / Pubmed: [Link] / Speaker: 방지윤
Our evaluations: ★★★

[2025.09.15] Microglia regulate GABAergic neurogenesis in prenatal human brain through IGF1. Yu et al. Nature. 2025
GABAergic neurons are essential cellular components of neural circuits. Their abundance and diversity have increased significantly in the human brain, contributing to the expanded cognitive capacity of humans1. However, the developmental mechanism underlying the extended production of GABAergic neurons in the human brain remains elusive. Here we uncovered the microglial regulation of the sustained proliferation of GABAergic progenitors and neuroblasts in the human medial ganglionic eminence (hMGE). We showed that microglia are preferentially distributed in the proliferating zone and identified insulin-like growth factor 1 (IGF1) and its receptor IGR1R as the predicted top ligand-receptor pair underlying microglia-progenitor communication in the prenatal hMGE. Using our newly developed neuroimmune hMGE organoids, which mimic the hMGE cytoarchitecture and developmental trajectory, we demonstrated that microglia-derived IGF1 promotes progenitor proliferation and production of GABAergic neurons. Conversely, IGF1-neutralizing antibodies and IGF1 knockout human embryonic stem-cell-induced microglia abolish the induced microglia-mediated progenitor proliferation. Together, these findings revealed a previously unappreciated role of microglia-derived IGF1 in promoting the proliferation of neural progenitors and the development of GABAergic neurons in the human brain.
Research group: Xianhua Piao Lab [Link] / Pubmed: [Link] / Speaker: 이진아
Our evaluations: ★★★

[2025.09.08] Microglia rescue neurons from aggregate-induced neuronal dysfunction and death through tunneling nanotubes. Scheiblich et al. Neuron. 2024
Microglia are crucial for maintaining brain health and neuron function. Here, we report that microglia establish connections with neurons using tunneling nanotubes (TNTs) in both physiological and pathological conditions. These TNTs facilitate the rapid exchange of organelles, vesicles, and proteins. In neurodegenerative diseases like Parkinson's and Alzheimer's disease, toxic aggregates of alpha-synuclein (α-syn) and tau accumulate within neurons. Our research demonstrates that microglia use TNTs to extract neurons from these aggregates, restoring neuronal health. Additionally, microglia share their healthy mitochondria with burdened neurons, reducing oxidative stress and normalizing gene expression. Disrupting mitochondrial function with antimycin A before TNT formation eliminates this neuroprotection. Moreover, co-culturing neurons with microglia and promoting TNT formation rescues suppressed neuronal activity caused by α-syn or tau aggregates. Notably, TNT-mediated aggregate transfer is compromised in microglia carrying Lrrk22(Gly2019Ser) or Trem2(T66M) and (R47H) mutations, suggesting a role in the pathology of these gene variants in neurodegenerative diseases.
Research group: Michael T. Heneka Lab [Link] / Pubmed: [Link] / Speaker: 김우석 (Han’s Lab)

[2025.08.19] Amyloid-β induces lipid droplet-mediated microglial dysfunction via the enzyme DGAT2 in Alzheimer's disease. Prakash et al., Immunity. 2025
Microglial phagocytosis genes have been linked to increased risk for Alzheimer's disease (AD), but the mechanisms translating genetic association to cellular dysfunction remain unknown. Here, we showed that microglia formed lipid droplets (LDs) upon amyloid-β (Aβ) exposure and that LD loads increased with proximity to amyloid plaques in brains from individuals with AD and the 5xFAD mouse model. LD-laden microglia exhibited defects in Aβ phagocytosis, and unbiased lipidomic analyses identified a parallel decrease in free fatty acids (FFAs) and increase in triacylglycerols (TGs) as the key metabolic transition underlying LD formation. Diacylglycerol O-acyltransferase 2 (DGAT2)-a key enzyme that converts FFAs to TGs-promoted microglial LD formation and was increased in mouse 5xFAD and human AD brains. Pharmacologically targeting DGAT2 improved microglial uptake of Aβ and reduced plaque load and neuronal damage in 5xFAD mice. These findings identify a lipid-mediated mechanism underlying microglial dysfunction that could become a therapeutic target for AD.
Research group: Gaurav Chopra Lab [Link] / Pubmed: [Link] / Speaker: Ruiying Ma (마서영) (Han’s Lab)

[2025.08.14] GABA-dependent microglial elimination of inhibitory synapses underlies neuronal hyperexcitability in epilepsy. Chen et al. Nat Neurosci. 2025
Neuronal hyperexcitability is a common pathophysiological feature of many neurological diseases. Neuron-glia interactions underlie this process but the detailed mechanisms remain unclear. Here, we reveal a critical role of microglia-mediated selective elimination of inhibitory synapses in driving neuronal hyperexcitability. In epileptic mice of both sexes, hyperactive inhibitory neurons directly activate surveilling microglia via GABAergic signaling. In response, these activated microglia preferentially phagocytose inhibitory synapses, disrupting the balance between excitatory and inhibitory synaptic transmission and amplifying network excitability. This feedback mechanism depends on both GABA-GABAB receptor-mediated microglial activation and complement C3-C3aR-mediated microglial engulfment of inhibitory synapses, as pharmacological or genetic blockage of both pathways effectively prevents inhibitory synapse loss and ameliorates seizure symptoms in mice. Additionally, putative cell-cell interaction analyses of brain tissues from males and females with temporal lobe epilepsy reveal that inhibitory neurons induce microglial phagocytic states and inhibitory synapse loss. Our findings demonstrate that inhibitory neurons can directly instruct microglial states to control inhibitory synaptic transmission through a feedback mechanism, leading to the development of neuronal hyperexcitability in temporal lobe epilepsy.
Research group: Chao Yan Lab / Pubmed: [Link] / Speaker: 오준용 (Han’s Lab)

[2025.08.05] Microglia replacement halts the progression of microgliopathy in mice and humans. Wu et al. Science. 2025
Colony-stimulating factor 1 receptor (CSF1R) is primarily expressed in microglia. Its monoallelic mutation causes CSF1R-associated microgliopathy (CAMP), a major form of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) and a fatal neurological disease without clinical cure. We developed mouse models harboring human hotspot mutations of CAMP and replaced CSF1R-deficient microglia with CSF1R-normal cells through microglia replacement by bone marrow transplantation (Mr BMT), which attenuated pathology in mice. We further demonstrated that, in the context of CSF1R deficiency, traditional bone marrow transplantation (tBMT) in ALSP functions similarly to Mr BMT, efficiently replacing microglia and reducing disease progression. We then replaced CSF1R-deficient microglia in eight patients by tBMT. The disease progression was halted during the 24-month follow-up. Together, microglia replacement corrects pathogenic mutations and halts disease progression in mice and humans.
Research group: Bo Peng Lab [Link] / Pubmed: [Link] / Speaker: Alex

[2025.07.29] APOE4 impairs the microglial response in Alzheimer's disease by inducing TGFβ-mediated checkpoints. Yin et al. Nat Immunol. 2023
The APOE4 allele is the strongest genetic risk factor for late-onset Alzheimer's disease (AD). The contribution of microglial APOE4 to AD pathogenesis is unknown, although APOE has the most enriched gene expression in neurodegenerative microglia (MGnD). Here, we show in mice and humans a negative role of microglial APOE4 in the induction of the MGnD response to neurodegeneration. Deletion of microglial APOE4 restores the MGnD phenotype associated with neuroprotection in P301S tau transgenic mice and decreases pathology in APP/PS1 mice. MGnD-astrocyte cross-talk associated with β-amyloid (Aβ) plaque encapsulation and clearance are mediated via LGALS3 signaling following microglial APOE4 deletion. In the brains of AD donors carrying the APOE4 allele, we found a sex-dependent reciprocal induction of AD risk factors associated with suppression of MGnD genes in females, including LGALS3, compared to individuals homozygous for the APOE3 allele. Mechanistically, APOE4-mediated induction of ITGB8-transforming growth factor-β (TGFβ) signaling impairs the MGnD response via upregulation of microglial homeostatic checkpoints, including Inpp5d, in mice. Deletion of Inpp5d in microglia restores MGnD-astrocyte cross-talk and facilitates plaque clearance in APP/PS1 mice. We identify the microglial APOE4-ITGB8-TGFβ pathway as a negative regulator of microglial response to AD pathology, and restoring the MGnD phenotype via blocking ITGB8-TGFβ signaling provides a promising therapeutic intervention for AD.
Research group: Oleg Butovsky Lab [Link] / Pubmed: [Link] / Speaker: 류가영 (Han’s Lab)

[2025.07.15] Microglia are not necessary for maintenance of blood-brain barrier properties in health, but PLX5622 alters brain endothelial cholesterol metabolism. Profaci et al. Neuron. 2024
Microglia, the resident immune cells of the central nervous system, are intimately involved in the brain's most basic processes, from pruning neural synapses during development to preventing excessive neuronal activity throughout life. Studies have reported both helpful and harmful roles for microglia at the blood-brain barrier (BBB) in the context of disease. However, less is known about microglia-endothelial cell interactions in the healthy brain. To investigate the role of microglia at a healthy BBB, we used the colony-stimulating factor 1 receptor (CSF1R) inhibitor PLX5622 to deplete microglia and analyzed the BBB ultrastructure, permeability, and transcriptome. Interestingly, we found that, despite their direct contact with endothelial cells, microglia are not necessary for the maintenance of BBB structure, function, or gene expression in the healthy brain. However, we found that PLX5622 treatment alters brain endothelial cholesterol metabolism. This effect was independent from microglial depletion, suggesting that PLX5622 has off-target effects on brain vasculature.
Research group: Richard Daneman Lab [Link] / Pubmed: [Link] / Speaker: 유용진
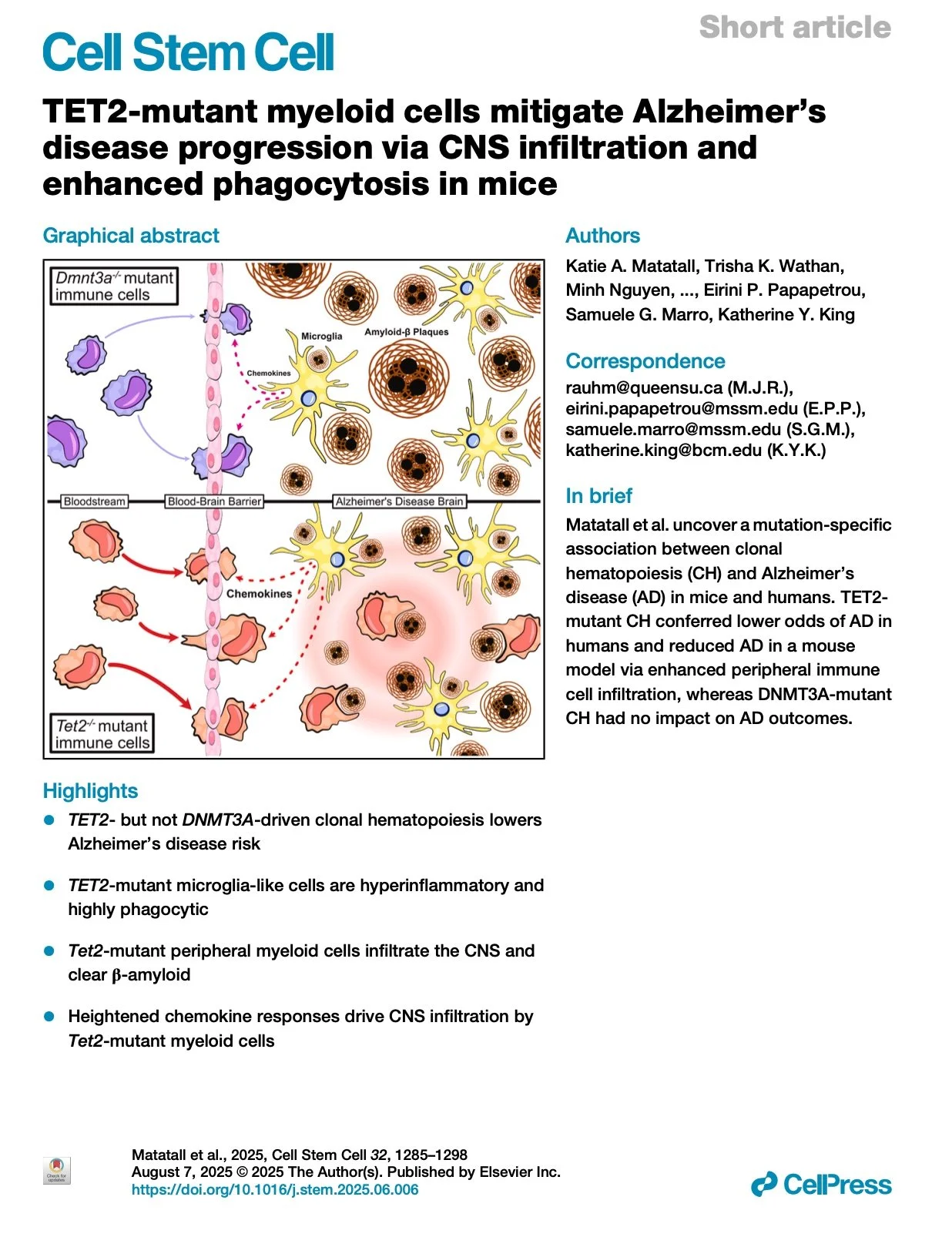
[2025.07.08] TET2-mutant myeloid cells mitigate Alzheimer’s disease progression via CNS infiltration and enhanced phagocytosis in mice. Matatall et al. Cell Stem Cell. 2025
Clonal hematopoiesis (CH) is associated with many age-related diseases, but its interaction with Alzheimer's disease (AD) remains unclear. Here, we show that TET2-mutant CH is associated with a 47% reduced risk of late-onset AD (LOAD) in the UK Biobank, whereas other drivers of CH do not confer protection. In a mouse model of AD, transplantation of Tet2-mutant bone marrow reduced cognitive decline and β-amyloid plaque formation, effects not observed with Dnmt3a-mutant marrow. Bone-marrow-derived microglia-like cells were detected at an increased rate in Tet2-mutant marrow recipients, and TET2-mutant human induced pluripotent stem cell (iPSC)-derived microglia were more phagocytic and hyperinflammatory than DNMT3A-mutant or wild-type microglia. Strikingly, single-cell RNA sequencing (scRNA-seq) revealed that macrophages and patrolling monocytes were increased in brains of mice transplanted with Tet2-mutant marrow in response to chemokine signaling. These studies reveal a TET2-specific protective effect of CH on AD pathogenesis mediated by peripheral myeloid cell infiltration.
Research group: Katherine Y King Lab [Link] / Pubmed: [Link] / Speaker: 방지윤

[2025.07.01] Microglia activation orchestrates CXCL10-mediated CD8+ T cell recruitment to promote aging-related white matter degeneration. Groh et al. Nat Neurosci. 2025
Aging is the major risk factor for neurodegeneration and is associated with structural and functional alterations in white matter. Myelin is particularly vulnerable to aging, resulting in white matter-associated microglia activation. Here we used pharmacological and genetic approaches to investigate microglial functions related to aging-associated changes in myelinated axons of mice. Our results reveal that maladaptive microglia activation promotes the accumulation of harmful CD8+ T cells, leading to the degeneration of myelinated axons and subsequent impairment of brain function and behavior. We characterize glial heterogeneity and aging-related changes in white matter by single-cell and spatial transcriptomics and reveal elaborate glial-immune interactions. Mechanistically, we show that the CXCL10-CXCR3 axis is crucial for the recruitment and retention of CD8+ T cells in aged white matter, where they exert pathogenic effects. Our results indicate that myelin-related microglia dysfunction promotes adaptive immune reactions in aging and identify putative targets to mitigate their detrimental impact.
Research group: Mikael Simons Lab [Link] / Pubmed: [Link] / Speaker: 이진아

[2025.06.24] Direct microglia replacement reveals pathologic and therapeutic contributions of brain macrophages to a monogenic neurological disease. Aisenberg et al. Immunity. 2025
Krabbe disease, also named globoid cell (GC) leukodystrophy (GLD) for its distinct lipid-laden macrophages, is a severe leukodystrophy caused by galactosylceramidase (GALC) mutations. Hematopoietic stem cell transplant (HSCT) ameliorates disease and is associated with central nervous system (CNS) engraftment of GALC+ donor macrophages. Yet, the role of macrophages in GLD pathophysiology and HSCT remains unclear. Using single-cell sequencing, we revealed early interferon response signatures that preceded progressively severe macrophage dyshomeostasis and identified a molecular signature of GCs, which we validated in human brain specimens. Genetic depletion and direct microglia replacement by CNS monocyte injection rapidly replaced >80% of endogenous microglia with healthy macrophages in the twitcher (GalcW355∗) mouse model of GLD. Perinatal microglia replacement completely normalized transcriptional signatures, rescued histopathology, and doubled average survival. Overall, we uncovered distinct forms of microglial dysfunction and evidence that direct, CNS-limited microglia replacement improves a monogenic neurodegenerative disease, identifying a promising therapeutic target.
Research group: F Chris Bennett Lab [Link] / Pubmed: [Link] / Speaker: 김우석 (Han’s Lab)

[2025.06.17] Immune checkpoint TIM-3 regulates microglia and Alzheimer's disease. Kimura et al. Nature. 2025
Microglia are the resident immune cells in the brain and have pivotal roles in neurodevelopment and neuroinflammation1,2. This study investigates the function of the immune-checkpoint molecule TIM-3 (encoded by HAVCR2) in microglia. TIM-3 was recently identified as a genetic risk factor for late-onset Alzheimer's disease3, and it can induce T cell exhaustion4. However, its specific function in brain microglia remains unclear. We demonstrate in mouse models that TGFβ signalling induces TIM-3 expression in microglia. In turn, TIM-3 interacts with SMAD2 and TGFBR2 through its carboxy-terminal tail, which enhances TGFβ signalling by promoting TGFBR-mediated SMAD2 phosphorylation, and this process maintains microglial homeostasis. Genetic deletion of Havcr2 in microglia leads to increased phagocytic activity and a gene-expression profile consistent with the neurodegenerative microglial phenotype (MGnD), also referred to as disease-associated microglia (DAM). Furthermore, microglia-targeted deletion of Havcr2 ameliorates cognitive impairment and reduces amyloid-β pathology in 5×FAD mice (a transgenic model of Alzheimer's disease). Single-nucleus RNA sequencing revealed a subpopulation of MGnD microglia in Havcr2-deficient 5×FAD mice characterized by increased pro-phagocytic and anti-inflammatory gene expression alongside reduced pro-inflammatory gene expression. These transcriptomic changes were corroborated by single-cell RNA sequencing data across most microglial clusters in Havcr2-deficient 5×FAD mice. Our findings reveal that TIM-3 mediates microglia homeostasis through TGFβ signalling and highlight the therapeutic potential of targeting microglial TIM-3 in Alzheimer's disease.
Research group: Vijay K Kuchroo Lab [Link] / Pubmed: [Link] / Speaker: Ruiying Ma (마서영) (Han’s Lab)
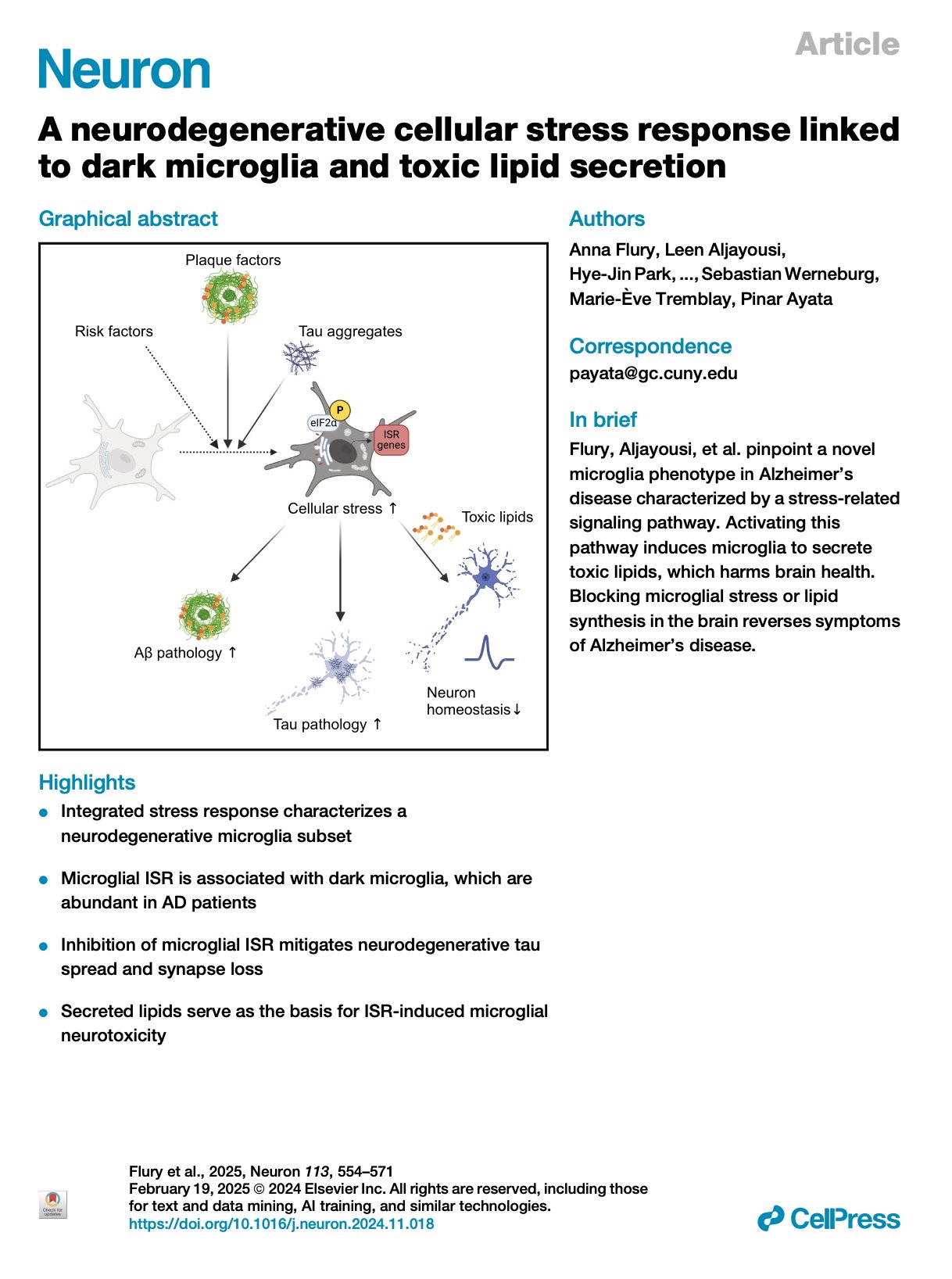
[2025.06.10] A neurodegenerative cellular stress response linked to dark microglia and toxic lipid secretion. Flury et al. Neuron. 2025
The brain's primary immune cells, microglia, are a leading causal cell type in Alzheimer's disease (AD). Yet, the mechanisms by which microglia can drive neurodegeneration remain unresolved. Here, we discover that a conserved stress signaling pathway, the integrated stress response (ISR), characterizes a microglia subset with neurodegenerative outcomes. Autonomous activation of ISR in microglia is sufficient to induce early features of the ultrastructurally distinct "dark microglia" linked to pathological synapse loss. In AD models, microglial ISR activation exacerbates neurodegenerative pathologies and synapse loss while its inhibition ameliorates them. Mechanistically, we present evidence that ISR activation promotes the secretion of toxic lipids by microglia, impairing neuron homeostasis and survival in vitro. Accordingly, pharmacological inhibition of ISR or lipid synthesis mitigates synapse loss in AD models. Our results demonstrate that microglial ISR activation represents a neurodegenerative phenotype, which may be sustained, at least in part, by the secretion of toxic lipids.
Research group: Pinar Ayata Lab [Link] / Pubmed: [Link] / Speaker: Alex

[2025.05.27] Somatic cancer driver mutations are enriched and associated with inflammatory states in Alzheimer's disease microglia. Huang et al. bioRxiv. 2024
Alzheimer's disease (AD) is an age-associated neurodegenerative disorder characterized by progressive neuronal loss and pathological accumulation of the misfolded proteins amyloid-β and tau1,2. Neuroinflammation mediated by microglia and brain-resident macrophages plays a crucial role in AD pathogenesis1-5, though the mechanisms by which age, genes, and other risk factors interact remain largely unknown. Somatic mutations accumulate with age and lead to clonal expansion of many cell types, contributing to cancer and many non-cancer diseases6,7. Here we studied somatic mutation in normal aged and AD brains by three orthogonal methods and in three independent AD cohorts. Analysis of bulk RNA sequencing data from 866 samples from different brain regions revealed significantly higher (~two-fold) overall burdens of somatic single-nucleotide variants (sSNVs) in AD brains compared to age-matched controls. Molecular-barcoded deep (>1000X) gene panel sequencing of 311 prefrontal cortex samples showed enrichment of sSNVs and somatic insertions and deletions (sIndels) in cancer driver genes in AD brain compared to control, with recurrent, and often multiple, mutations in genes implicated in clonal hematopoiesis (CH)8,9. Pathogenic sSNVs were enriched in CSF1R+ microglia of AD brains, and the high proportion of microglia (up to 40%) carrying some sSNVs in cancer driver genes suggests mutation-driven microglial clonal expansion (MiCE). Analysis of single-nucleus RNA sequencing (snRNAseq) from temporal neocortex of 62 additional AD cases and controls exhibited nominally increased mosaic chromosomal alterations (mCAs) associated with CH10,11. Microglia carrying mCA showed upregulated pro-inflammatory genes, resembling the transcriptomic features of disease-associated microglia (DAM) in AD. Our results suggest that somatic driver mutations in microglia are common with normal aging but further enriched in AD brain, driving MiCE with inflammatory and DAM signatures. Our findings provide the first insights into microglial clonal dynamics in AD and identify potential new approaches to AD diagnosis and therapy.
Research group: Christopher A Walsh Lab [Link] / Pubmed: [Link] / Speaker: 류가영 (Han’s Lab)

[2025.05.13] Neuronal somatic mutations are increased in multiple sclerosis lesions. Motyer et al. Nat Neurosci. 2025
Neuroinflammation underpins neurodegeneration and clinical progression in multiple sclerosis (MS), but knowledge of processes linking these disease mechanisms remains incomplete. Here we investigated somatic single-nucleotide variants (sSNVs) in the genomes of 106 single neurons from post-mortem brain tissue of ten MS cases and 16 controls to determine whether somatic mutagenesis is involved. We observed an increase of 43.9 sSNVs per year in neurons from chronic MS lesions, a 2.5 times faster rate than in neurons from normal-appearing MS and control tissues. This difference was equivalent to 1,291 excess sSNVs in lesion neurons at 70 years of age compared to controls. We performed mutational signature analysis to investigate mechanisms underlying neuronal sSNVs and identified a signature characteristic of lesions with a strong, age-associated contribution to sSNV counts. This research suggests that neuroinflammation is mutagenic in the MS brain, potentially contributing to disease progression.
Research group: Justin P. Rubio Lab [Link] / Pubmed: [Link] / Speaker: 유용진

[2025.04.15] Brain-Engrafted Monocyte-derived Macrophages from Blood and Skull-Bone Marrow Exhibit Distinct Identities from Microglia. Du et al. bioRxiv. 2024
Microglia are thought to originate exclusively from primitive macrophage progenitors in the yolk sac (YS) and to persist throughout life without much contribution from definitive hematopoiesis. Here, using lineage tracing, pharmacological manipulation, and RNA-sequencing, we elucidated the presence and characteristics of monocyte-derived macrophages (MDMs) in the brain parenchyma at baseline and during microglia repopulation, and defined the core transcriptional signatures of brain-engrafted MDMs. Lineage tracing mouse models revealed that MDMs transiently express CD206 during brain engraftment as CD206 + microglia precursors in the YS. We found that brain-engrafted MDMs exhibit transcriptional and epigenetic characteristics akin to meningeal macrophages, likely due to environmental imprinting within the meningeal space. Utilizing parabiosis and skull transplantation, we demonstrated that monocytes from both peripheral blood and skull bone marrow can repopulate microglia-depleted brains. Our results reveal the heterogeneous origins and cellular dynamics of brain parenchymal macrophages at baseline and in models of microglia depletion.
Research group: Jonathan Kipnis Lab [Link] / Pubmed: [Link] / Speaker: 방지윤

[2025.03.18] Propagation of neuronal micronuclei regulates microglial characteristics. Yano et al. Nat Neurosci. 2025
Microglia-resident immune cells in the central nervous system-undergo morphological and functional changes in response to signals from the local environment and mature into various homeostatic states. However, niche signals underlying microglial differentiation and maturation remain unknown. Here, we show that neuronal micronuclei (MN) transfer to microglia, which is followed by changing microglial characteristics during the postnatal period. Neurons passing through a dense region of the developing neocortex give rise to MN and release them into the extracellular space, before being incorporated into microglia and inducing morphological changes. Two-photon imaging analyses have revealed that microglia incorporating MN tend to slowly retract their processes. Loss of the cGAS gene alleviates effects on micronucleus-dependent morphological changes. Neuronal MN-harboring microglia also exhibit unique transcriptome signatures. These results demonstrate that neuronal MN serve as niche signals that transform microglia, and provide a potential mechanism for regulation of microglial characteristics in the early postnatal neocortex.
Research group: Fuminori Tsuruta Lab [Link] / Pubmed: [Link] / Speaker: 이진아
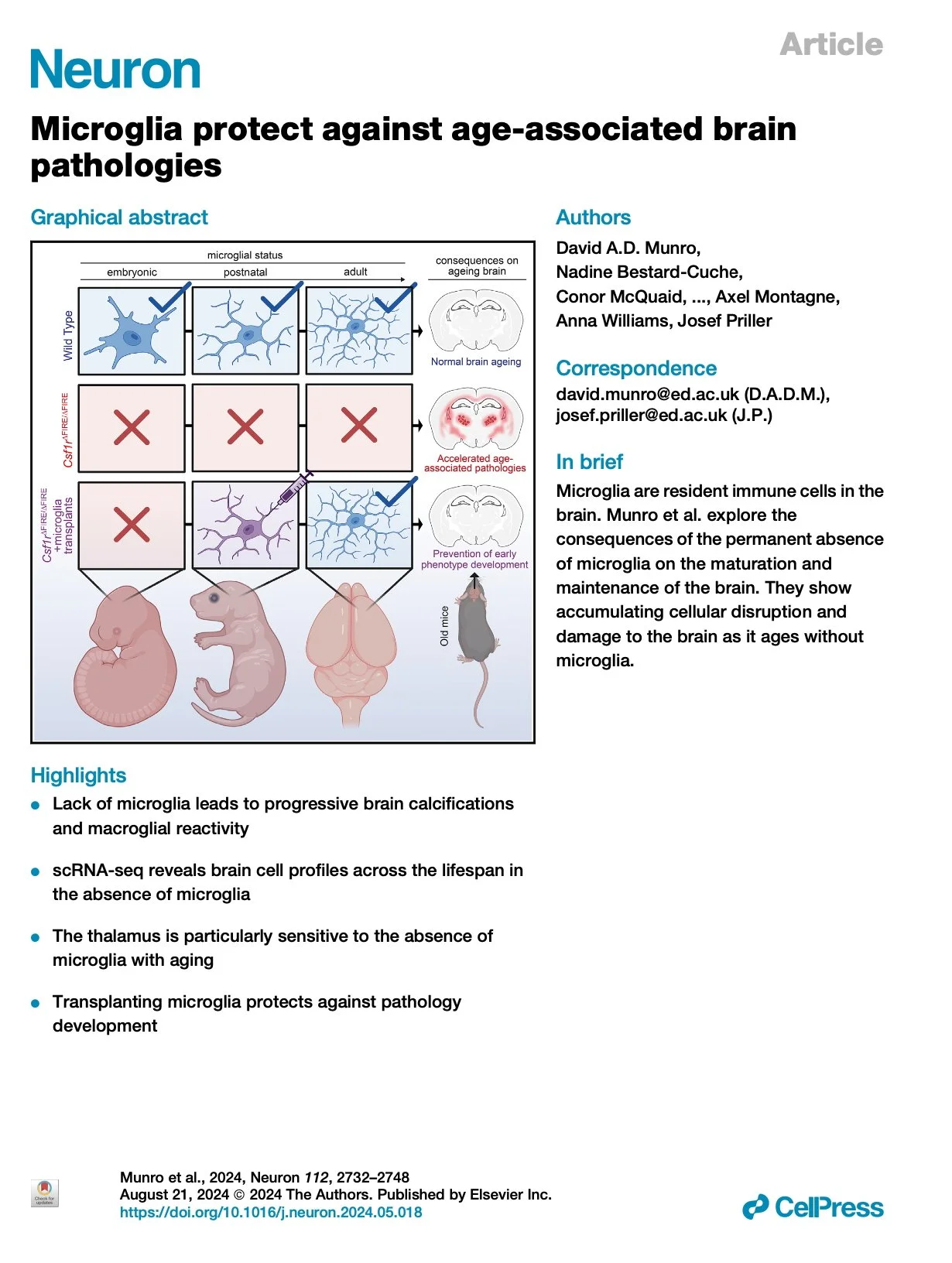
[2025.03.04] Microglia protect against age-associated brain pathologies. Munro et al. Neuron. 2024
Microglia are brain-resident macrophages that contribute to central nervous system (CNS) development, maturation, and preservation. Here, we examine the consequences of permanent microglial deficiencies on brain aging using the Csf1rΔFIRE/ΔFIRE mouse model. In juvenile Csf1rΔFIRE/ΔFIRE mice, we show that microglia are dispensable for the transcriptomic maturation of other brain cell types. By contrast, with advancing age, pathologies accumulate in Csf1rΔFIRE/ΔFIRE brains, macroglia become increasingly dysregulated, and white matter integrity declines, mimicking many pathological features of human CSF1R-related leukoencephalopathy. The thalamus is particularly vulnerable to neuropathological changes in the absence of microglia, with atrophy, neuron loss, vascular alterations, macroglial dysregulation, and severe tissue calcification. We show that populating Csf1rΔFIRE/ΔFIRE brains with wild-type microglia protects against many of these pathological changes. Together with the accompanying study by Chadarevian and colleagues1, our results indicate that the lifelong absence of microglia results in an age-related neurodegenerative condition that can be counteracted via transplantation of healthy microglia.
Research group: Josef Priller Lab [Link] / Pubmed: [Link] / Speaker: 김우석 (Han’s Lab)

[2025.02.11] Typical development of synaptic and neuronal properties can proceed without microglia in the cortex and thalamus. O'Keeffe et al. Nat Neurosci. 2025
Brain-resident macrophages, microglia, have been proposed to have an active role in synaptic refinement and maturation, influencing plasticity and circuit-level connectivity. Here we show that several neurodevelopmental processes previously attributed to microglia can proceed without them. Using a genetically modified mouse that lacks microglia (Csf1r∆FIRE/∆FIRE), we find that intrinsic properties, synapse number and synaptic maturation are largely normal in the hippocampal CA1 region and somatosensory cortex at stages where microglia have been implicated. Seizure susceptibility and hippocampal-prefrontal cortex coherence in awake behaving animals, processes that are disrupted in mice deficient in microglia-enriched genes, are also normal. Similarly, eye-specific segregation of inputs into the lateral geniculate nucleus proceeds normally in the absence of microglia. Single-cell and single-nucleus transcriptomic analyses of neurons and astrocytes did not uncover any substantial perturbation caused by microglial absence. Thus, the brain possesses remarkable adaptability to execute developmental synaptic refinement, maturation and connectivity in the absence of microglia.
Research group: Giles E Hardingham Lab [Link] / Pubmed: [Link] / Speaker: Ruiying Ma (마서영) (Han’s Lab)
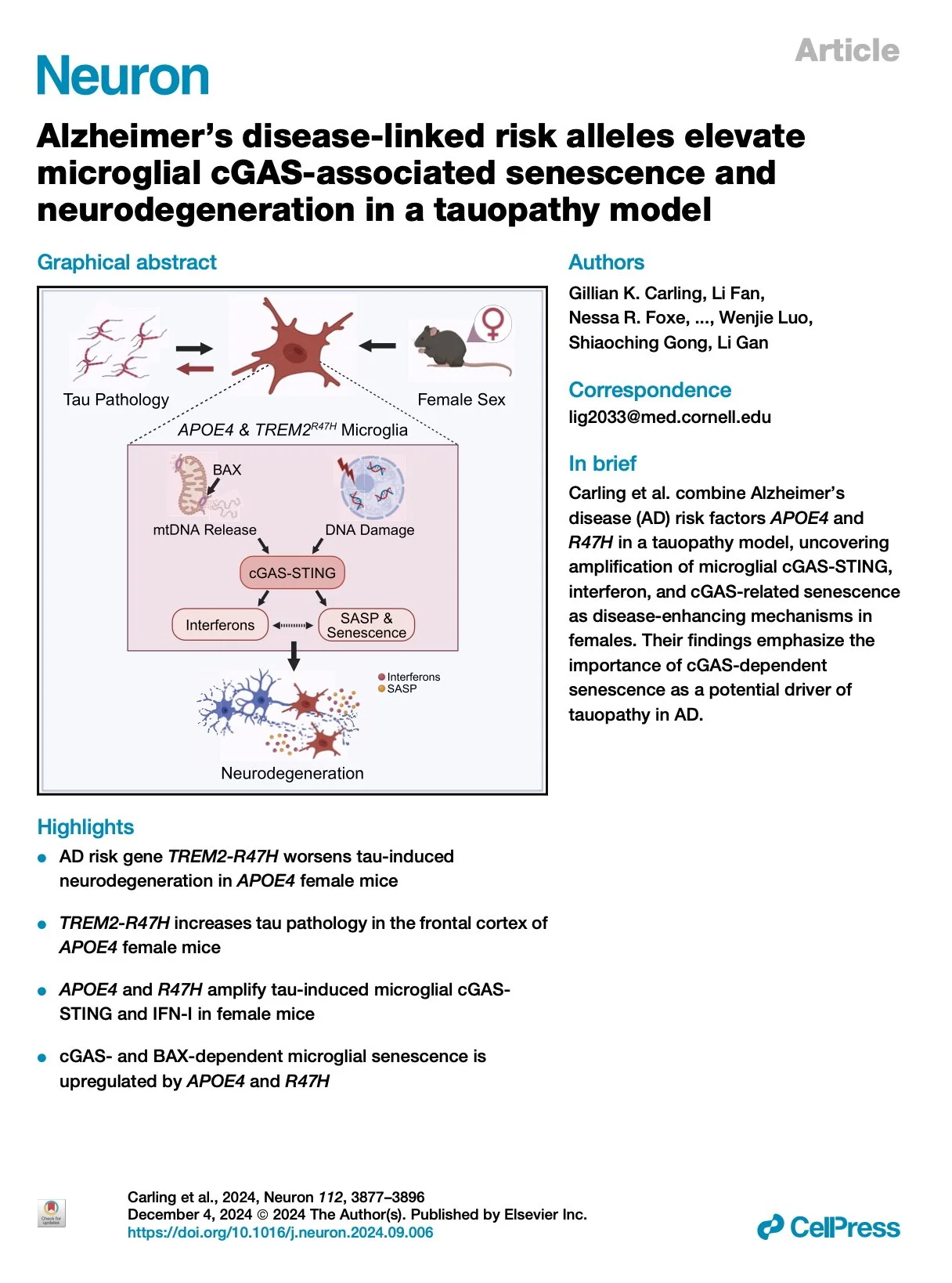
[2025.01.07] Alzheimer's disease-linked risk alleles elevate microglial cGAS-associated senescence and neurodegeneration in a tauopathy model. Carling et al. Neuron. 2024
The strongest risk factors for late-onset sporadic Alzheimer's disease (AD) include the ε4 allele of apolipoprotein E (APOE), the R47H variant of triggering receptor expressed on myeloid cells 2 (TREM2), and female sex. Here, we combine APOE4 and TREM2R47H (R47H) in female P301S tauopathy mice to identify the pathways activated when AD risk is the strongest, thereby highlighting detrimental disease mechanisms. We find that R47H induces neurodegeneration in 9- to 10-month-old female APOE4 tauopathy mice. The combination of APOE4 and R47H (APOE4-R47H) worsened hyperphosphorylated tau pathology in the frontal cortex and amplified tauopathy-induced microglial cyclic guanosine monophosphate (GMP)-AMP synthase (cGAS)-stimulator of interferon genes (STING) signaling and downstream interferon response. APOE4-R47H microglia displayed cGAS- and BAX-dependent upregulation of senescence, showing association between neurotoxic signatures and implicating mitochondrial permeabilization in pathogenesis. By uncovering pathways enhanced by the strongest AD risk factors, our study points to cGAS-STING signaling and associated microglial senescence as potential drivers of AD risk.
Research group: Li Gan Lab [Link] / Pubmed: [Link] / Speaker: Alex
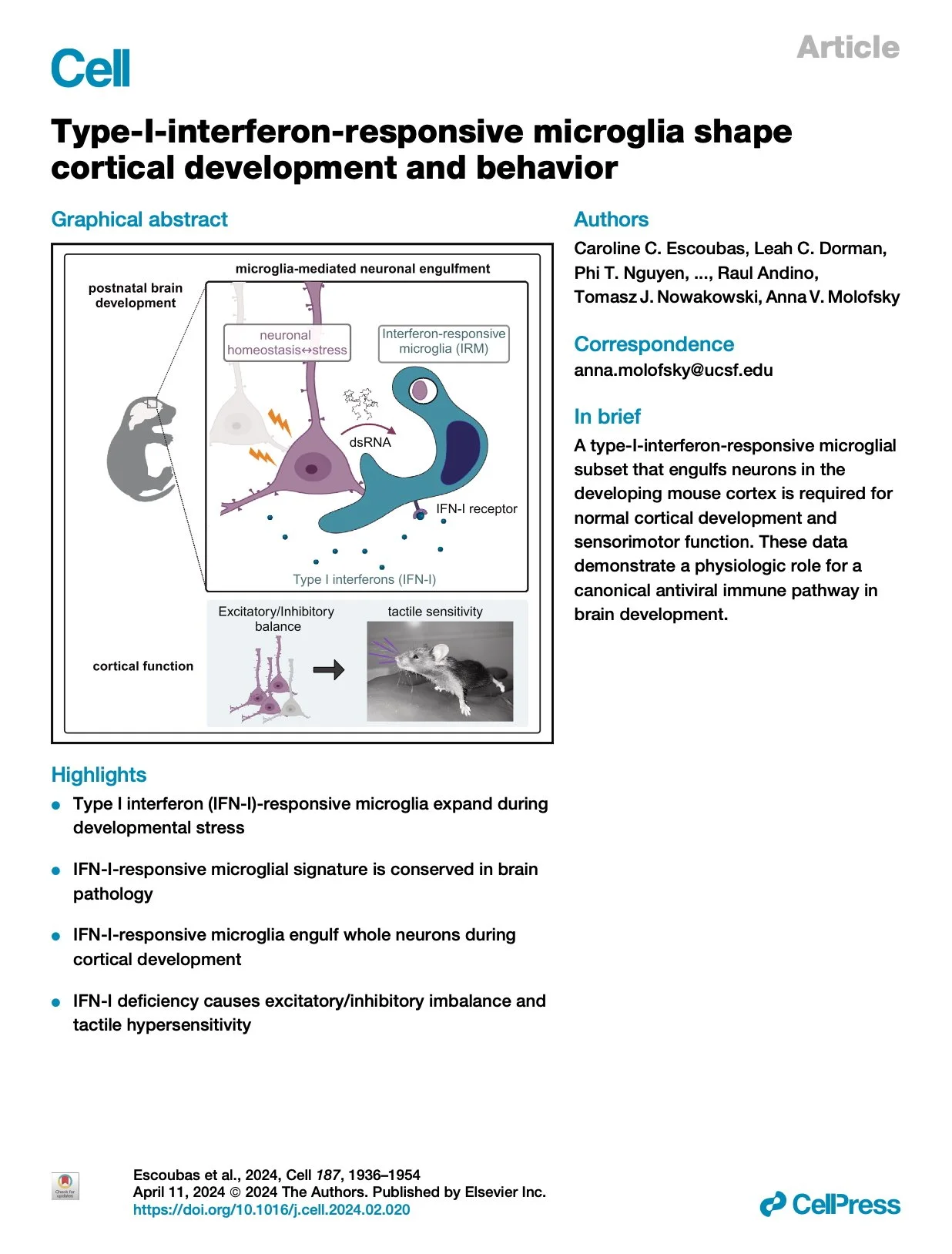
[2024.12.20] Type-I-interferon-responsive microglia shape cortical development and behavior. Escoubas et al. Cell. 2024
Microglia are brain-resident macrophages that shape neural circuit development and are implicated in neurodevelopmental diseases. Multiple microglial transcriptional states have been defined, but their functional significance is unclear. Here, we identify a type I interferon (IFN-I)-responsive microglial state in the developing somatosensory cortex (postnatal day 5) that is actively engulfing whole neurons. This population expands during cortical remodeling induced by partial whisker deprivation. Global or microglial-specific loss of the IFN-I receptor resulted in microglia with phagolysosomal dysfunction and an accumulation of neurons with nuclear DNA damage. IFN-I gain of function increased neuronal engulfment by microglia in both mouse and zebrafish and restricted the accumulation of DNA-damaged neurons. Finally, IFN-I deficiency resulted in excess cortical excitatory neurons and tactile hypersensitivity. These data define a role for neuron-engulfing microglia during a critical window of brain development and reveal homeostatic functions of a canonical antiviral signaling pathway in the brain.
Research group: Anna V Molofsky Lab [Link] / Pubmed: [Link] / Speaker: 유용진